


原子三维洞察Pd2Sn纳米合金的晶相效应
晶相是决定晶体材料原子排列方式的基本结构要素。对于纳米催化剂而言,不同晶相的本征结构对称性决定了其表面组成、原子排列、几何构型及配位环境,从而显著影响催化剂的整体性能。尤其在纳米尺度下,具有独特原子构型的非典型相结构的存在,为设计高性能纳米催化剂提供了理想平台。然而,由于表面弛豫与重构效应,准确阐释不同晶相纳米催化剂的化学反应性与表面原子排列之间的关系仍存在巨大挑战。近期,邢献然教授团队采用原子对分布函数(PDF)与反向蒙特卡洛(RMC)模拟相结合的方法,实现了对Pd2Sn纳米合金在乙炔加氢反应中晶相效应的原子三维解密,准确揭示出六方相Pd2Sn纳米晶体表面具有较低Pd-Pd配位数且被Sn原子高度隔离的Pd位点是乙炔选择性加氢的关键。相关工作发表在期刊ACS Catalysis上。
图 1 原文和摘要
作者团队通过精细调控热处理条件,成功实现了Pd2Sn纳米催化剂从正交相向六方相的晶体结构转变,使其在乙炔加氢反应中乙烯选择性显著提升至93.7%,该性能优于大多数已报道的先进催化剂。EXAFS和CO原位红外光谱的分析结果表明,六方相Pd2Sn纳米催化剂具有较低的平均Pd–Pd配位数。进一步,借助PDF和RMC相结合的方法,研究团队揭示了正交和六方Pd2Sn纳米晶的三维原子排布和表面Pd位点的分割度。定量分析结果显示,六方相Pd2Sn纳米晶表面Pd位点的分割度主要集中在0.5–1.25范围内,而正交相样品的分割度则多分布于1.25–1.75之间,表明六方相表面Pd位点具有更高的分散性和孤立特征。密度泛函理论(DFT)计算从机理上证实,乙炔的高选择性加氢源于六方相Pd2Sn独特的表面Pd局域配位结构及其Pd 4d与Sn 5p轨道间的适度杂化,该协同作用优化了反应中间体的吸脱附行为。
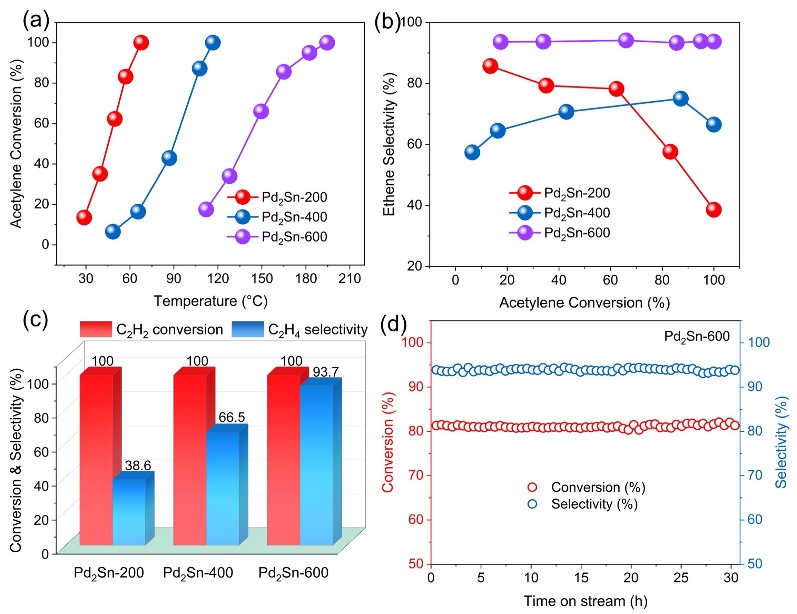
图2 Pd2Sn纳米催化剂的乙炔加氢性能
综上,该研究通过EXAFS、PDF和RMC等局域结构研究方法,实现了对乙炔加氢反应中Pd2Sn纳米合金晶相效应的三维原子尺度洞察,准确识别出高效选择性加氢活性位点。本研究为精确揭示活性位点的空间分布和精细调整活性位点的原子结构以提高纳米材料的催化性能提供了一个范例。
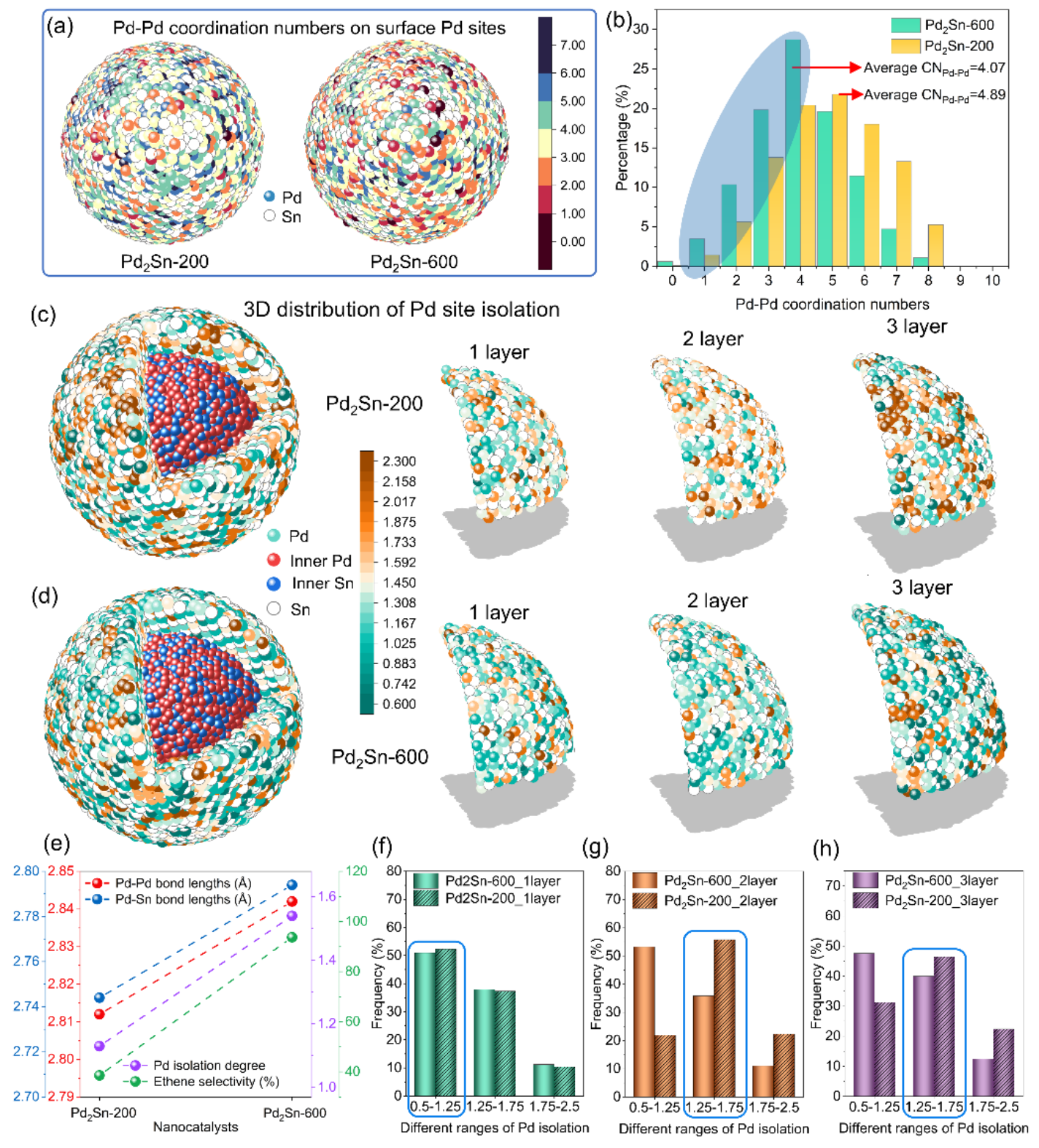
图3 不同晶相Pd2Sn纳米晶的三维原子结构和表面配位环境
该成果以"Active Site Isolation in Pd2Sn Nanocatalysts via Crystal Phase to Enhance Hydrogenation Selectivity "为题,近期发表在《美国化学会催化》(ACS Catalysis)。论文第一作者为北京科技大学固体化学研究所博士生薛凡和籍伟花,通讯作者为固体化学研究所邢献然教授和李强副教授。该工作得到国家自然科学基金的资助。论文链接: https://pubs.acs.org/doi/10.1021/acscatal.5c02460.