


PdCu纳米合金中乙炔高效半加氢活性位点原子尺度解析
准确地揭示表面活性位点三维原子结构对于高性能催化剂合理设计及催化机理洞察来说至关重要但极具挑战性。北京科技大学固体化学研究所邢献然教授团队采用原子对分布函数(PDF)和反向蒙特卡罗模拟(RMC)相结合方法,成功揭示了PdCu纳米合金中活性位点Pd的三维空间分布和近邻配位环境。通过对原子空间排布精细调控,Pd34Cu66纳米合金在乙炔完全转化时获得了98%超高乙烯选择性,优于目前报道的大多数先进催化剂。三维原子结构的定量解析表明,Pd34Cu66纳米合金表面分布着大量Pd-Pd配位数为3的活性位点,对乙炔高效半加氢起着决定性作用。这一发现不仅为从原子水平上指导双金属纳米催化剂的精确设计开辟了道路,而且为解析活性位点的空间结构提供了一种方法。
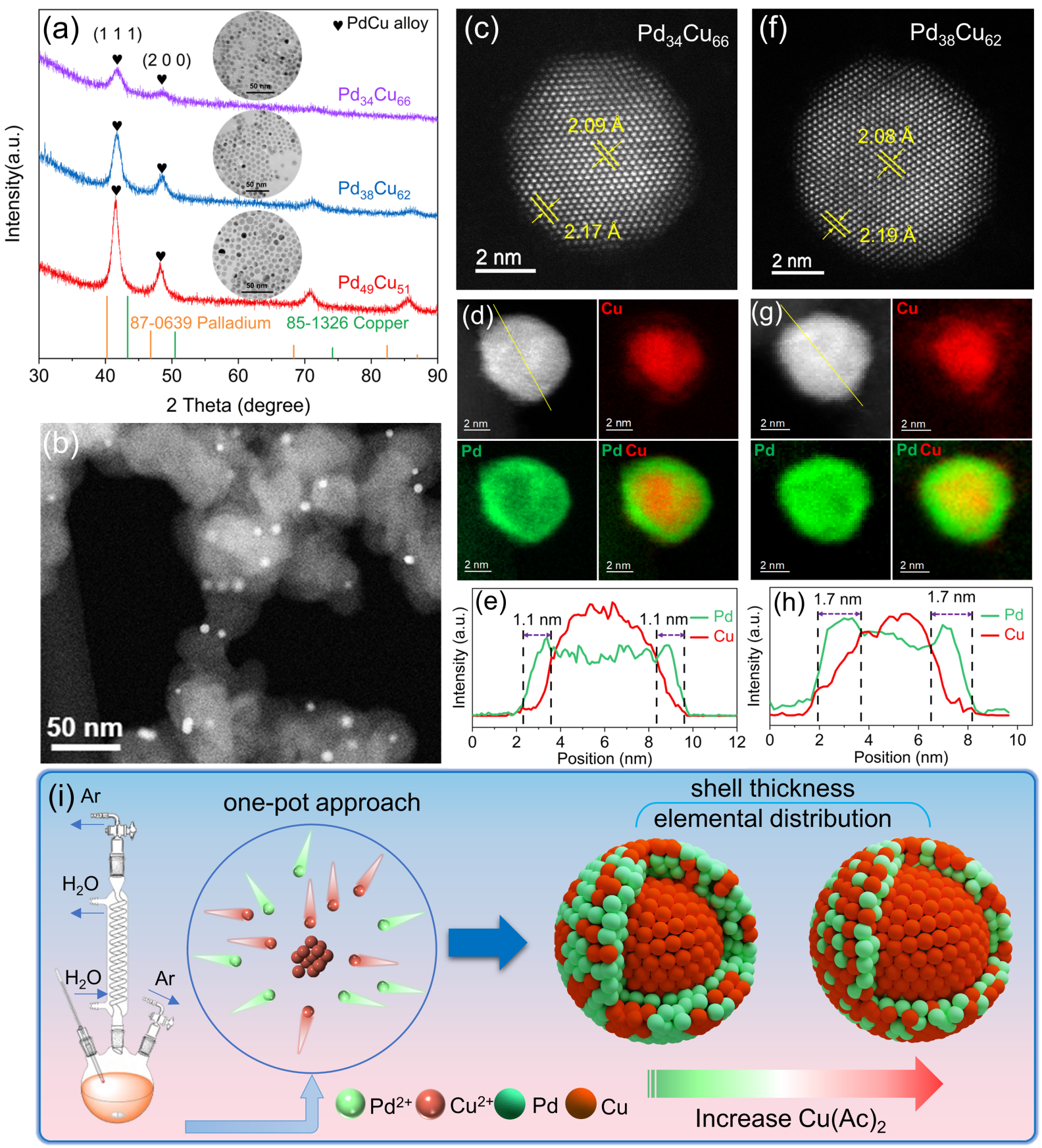
图 1 (a) 不同组分PdCu纳米晶的XRD谱图; (b) Pd34Cu66/C纳米催化剂的TEM图像;(c-e) Pd34Cu66 纳米晶的HAADF-STEM图像、电子能量损失谱(EELS)的元素面扫图以及元素线扫图;(f-h)Pd38Cu62 纳米晶的HAADF-STEM图像、电子能量损失谱(EELS)的元素面扫图以及元素线扫图;(i) Pd-Cu 纳米合金的元素偏析示意图
本研究采用简单的一锅法,利用Cu原子择优形核的特点,构建了PdCu合金壳,并且通过微调铜前驱体的含量,有效调节了壳层厚度和表面元素分布(如图1所示)。以乙炔加氢作为探针反应来研究表面局域结构与催化性能之间的关系。性能测试结果表明,在乙炔完全转化条件下,Pd34Cu66/C纳米催化剂表现出98%的超高乙烯选择性,远高于平均结构相似的Pd38Cu62/C催化剂,也同样优于之前报道的大多数相关催化剂。
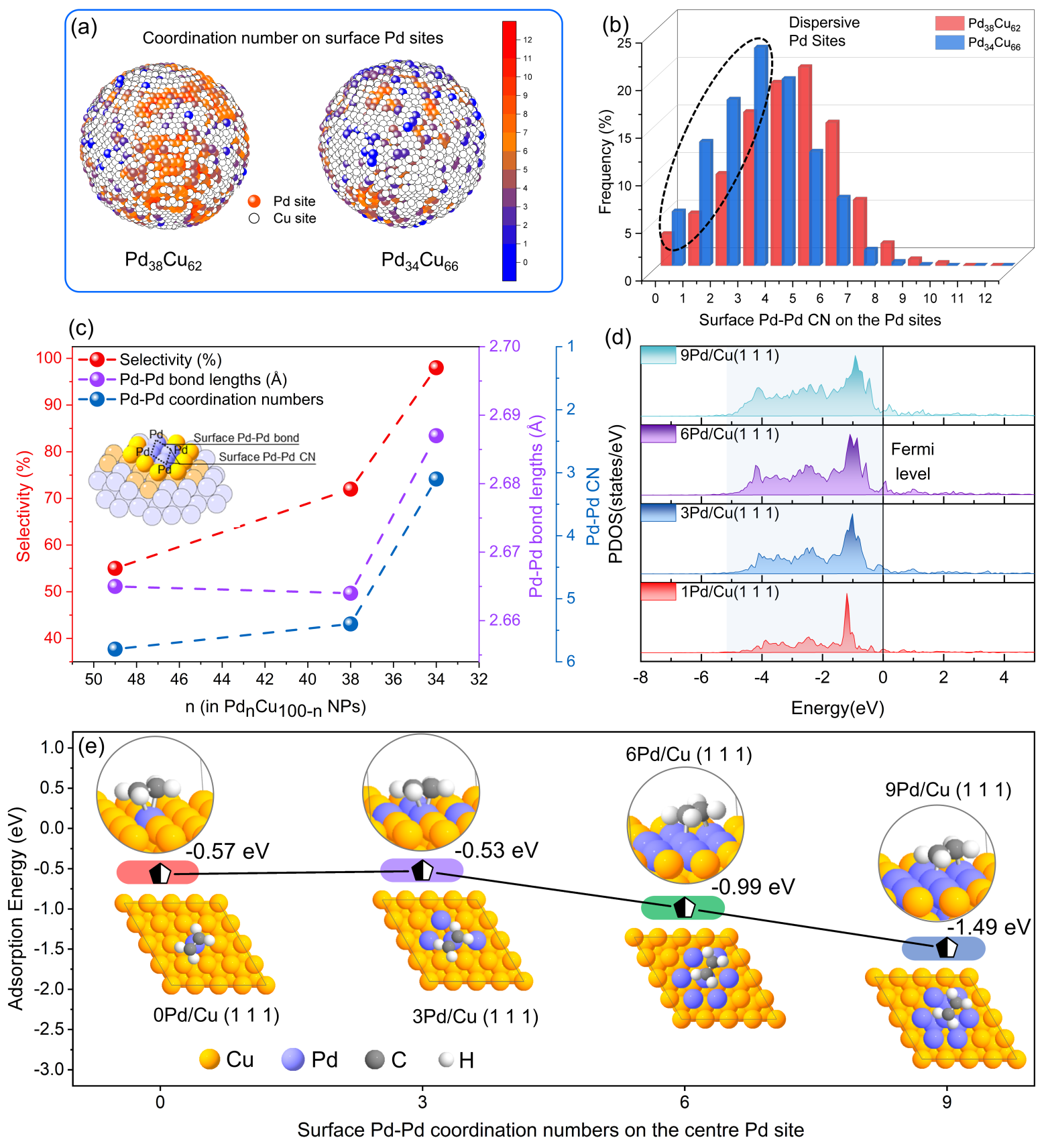
图2 (a)PdCu纳米晶中最近邻Pd-Pd配位数的三维分布;(b)PdCu纳米晶体表面Pd-Pd配位数分布;(c)乙烯选择性与PdCu纳米晶表面Pd-Pd原子对距离和Pd-Pd配位数的关系;(d)Pd的d轨道投影态密度图(e)乙烯吸附能与表面Pd-Pd配位数的关系
利用反向蒙特卡洛模拟(RMC)的方法对原子对分布函数(PDF)数据进行拟合,获得了PdCu纳米催化剂的三维原子结构和表面活性位的局域配位结构。和Pd38Cu62纳米合金相比,Pd34Cu66表面分布着大量Pd-Pd配位数为3的离散型Pd位点。DFT计算结果表明,活性位点Pd在Pd34Cu66合金表面的适当离散分布导致了乙烯的最佳吸附,有助于实现乙炔高效半加氢(如图2所示)。这项工作为活性位点的三维分布的解析和Pd基双金属的合理设计提供了重要的指导。
该成果以“PdCu纳米合金中乙炔高效半加氢活性位点解析”(Decoding Active Sites for Highly Efficient Semihydrogenation of Acetylene in Palladium–Copper Nanoalloys)为题,近期发表在《纳米通讯》(Nano Lett. 2024, 24, 6269-6277)。论文的第一作者为固体化学所博士生薛凡,共同通讯作者为北京科技大学固体化学所邢献然教授、李强副教授。
论文链接:https://pubs.acs.org/doi/full/10.1021/acs.nanolett.4c00941
该工作与国内相关单位合作完成,得到国家自然科学基金、科技部国家重点研发计划等项目资助。